

Cystic fibrosis (CF) is a genetic condition that affects many organs in the body: especially the lungs, pancreas and sweat glands. People affected by CF produce abnormally thick, sticky mucus, particularly in the lungs and digestive system. Intelligence is not affected in people with CF.
While it is normal to have mucus lining the organs of the respiratory, digestive, and reproductive systems, in people with CF this mucus is thick and sticky which can trap infections in the lungs leading to progressive lung damage. Digestion is affected as the body cannot extract nutrients from the normal diet, this can be improved by taking enzyme supplements however, commonly people with CF struggle with maintaining healthy weight despite a good appetite and balanced diet. FInally, the sweat glands also secrete sweat that is very high in salt, thereby depleting the body of this important substance.
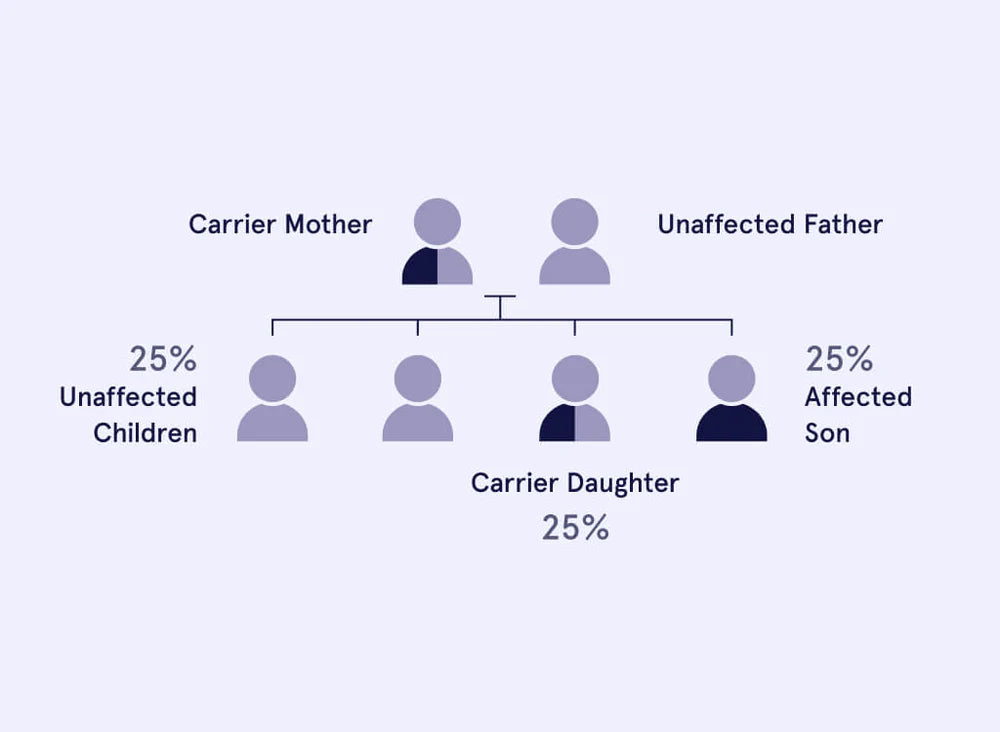
The severity of symptoms varies from person to person, even among individuals in the same family and/or with the same mutations. Males with CF may also appear as infertile. This is because most men with CF are missing the tubes that transport sperm from the testicles out through the penis - this is called congenital absence of the vas deferens (CAVD). Men with CF still produce normal mature sperm and thus infertility can be overcome using IVF.
How frequently is it carried by individuals (Carrier Frequency)?
Ethnicity influences the chances of carrying the Cystic Fibrosis mutation. Cystic fibrosis affects most commonly people who are of Northern European or UK descent, fairly frequently people whose ancestry is Southern European and Middle Eastern populations, but is rare where the ancestry is Asian.
When and how is it diagnosed?
Before a child is born
Pre-pregnancy screening can identify couples at risk of having a child affected with CF. It is important to know that CF is not part of standard first trimester genetic tests and may sometimes be found by chance during routine second trimester ultrasounds. If a pregnancy is known to be at risk a test may be requested and/or offered to diagnose the genetic status of the pregnancy. This can be done as early as the 11th week of pregnancy.
At birth:
Most cases of CF are diagnosed following Newborn screening which is a heel-prick blood test offered to all newborns internationally and aims to identify babies that have an abnormal level of certain chemicals in their blood.
Testing of people thought to be at risk may include a sweat-test to measure the concentration of salt in the sweat, and then by genetic testing of the CFTR gene. More than 1000 spelling mistakes (mutations) have been shown to affect the function of the CFTR gene and thus genetic testing is the ultimate tool in diagnosis.
In general, individuals with two classic mutations are more likely to have a severe form of the disease including problems with the pancreas, while individuals with one classic and one non-classic or individuals with two non-classic mutations are more likely to have a milder form of the condition and may avoid problems with the pancreas. It is important however to remember that not all people with the same mutation will have the same severity or prognosis.
Most people with CF experience breathing problems and frequent lung infections that lead to permanent lung damage such as scarring (fibrosis) and sac-like growths (cysts). The pancreas, an organ that produces insulin and digestive enzymes, is often affected by CF. The sticky mucus caused by CF can block ducts which ferry enzymes from the pancreas to the rest of the body, resulting in problems such as diarrhea, malnutrition, and poor growth. Infertility, particularly in men, and delayed puberty are also common among people with cystic fibrosis.
How can it be treated?
There is no treatment that addresses the cause of CF, but there are many options to treat the symptoms it produces. Daily physiotherapy in combination with prescription medication can help clear mucus from the lungs, improving lung function and reducing the risk of infection. As respiratory infections occur, physicians typically prescribe antibiotics. SOmetimes people will require hospital admissions for health support.
Physicians will also monitor the digestive system to ensure that the person is getting proper nutrition. Enzymes or vitamin supplements may be prescribed. Both the respiratory and digestive systems of a person with CF must be monitored regularly by his or her medical team.
Surgery may be needed to correct certain problems caused by CF. Lung transplants are an option for some people. More recently genetic therapies have shown to improve the quality of life of people with CF.
What is the prognosis for a person with Cystic Fibrosis?
There is no cure for CF. However, thanks to improved treatments and a better understanding of the condition, the average life expectancy for people with CF who live to adulthood is 35 years. Children born with CF today who receive early treatment may live even longer.
Resources




