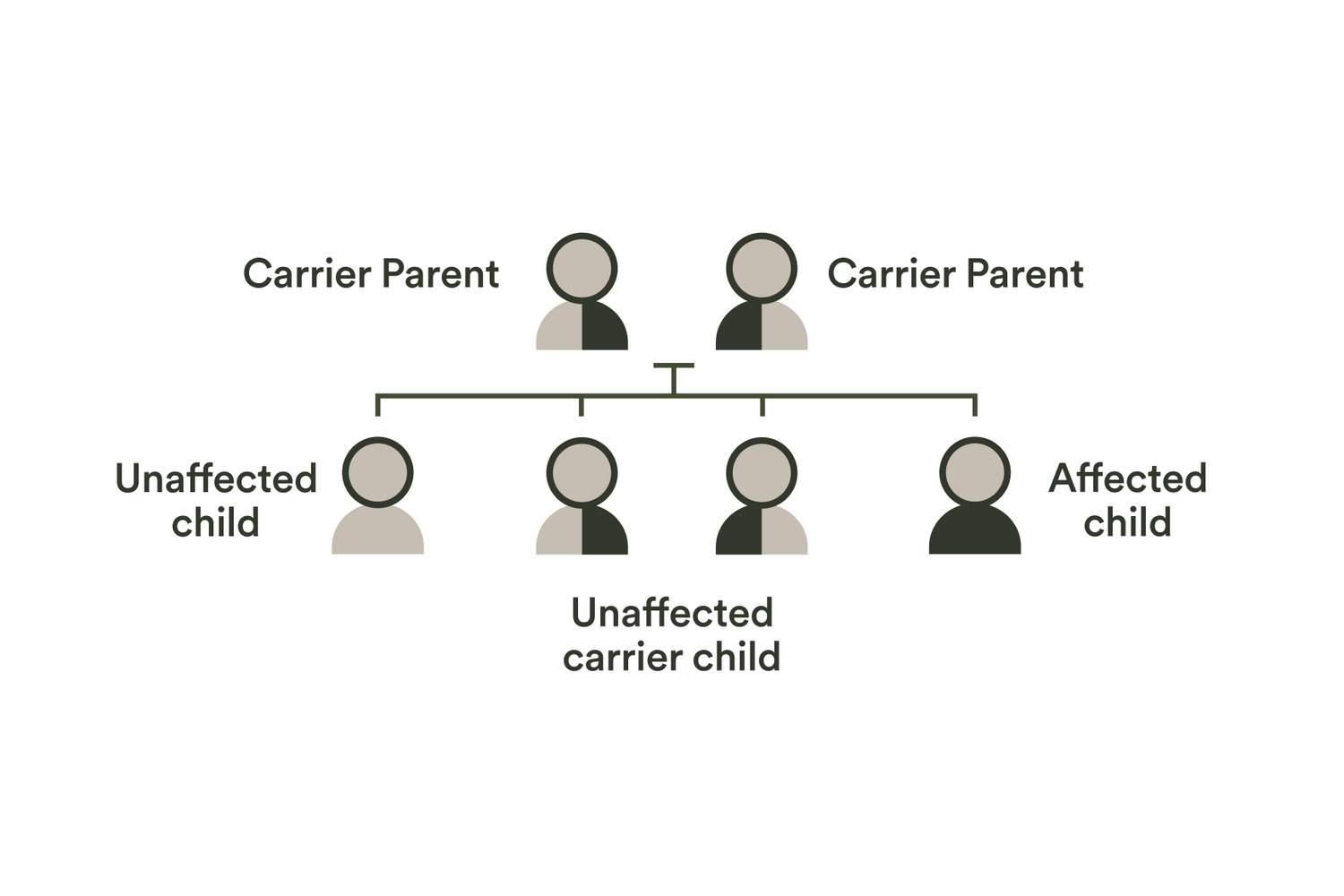
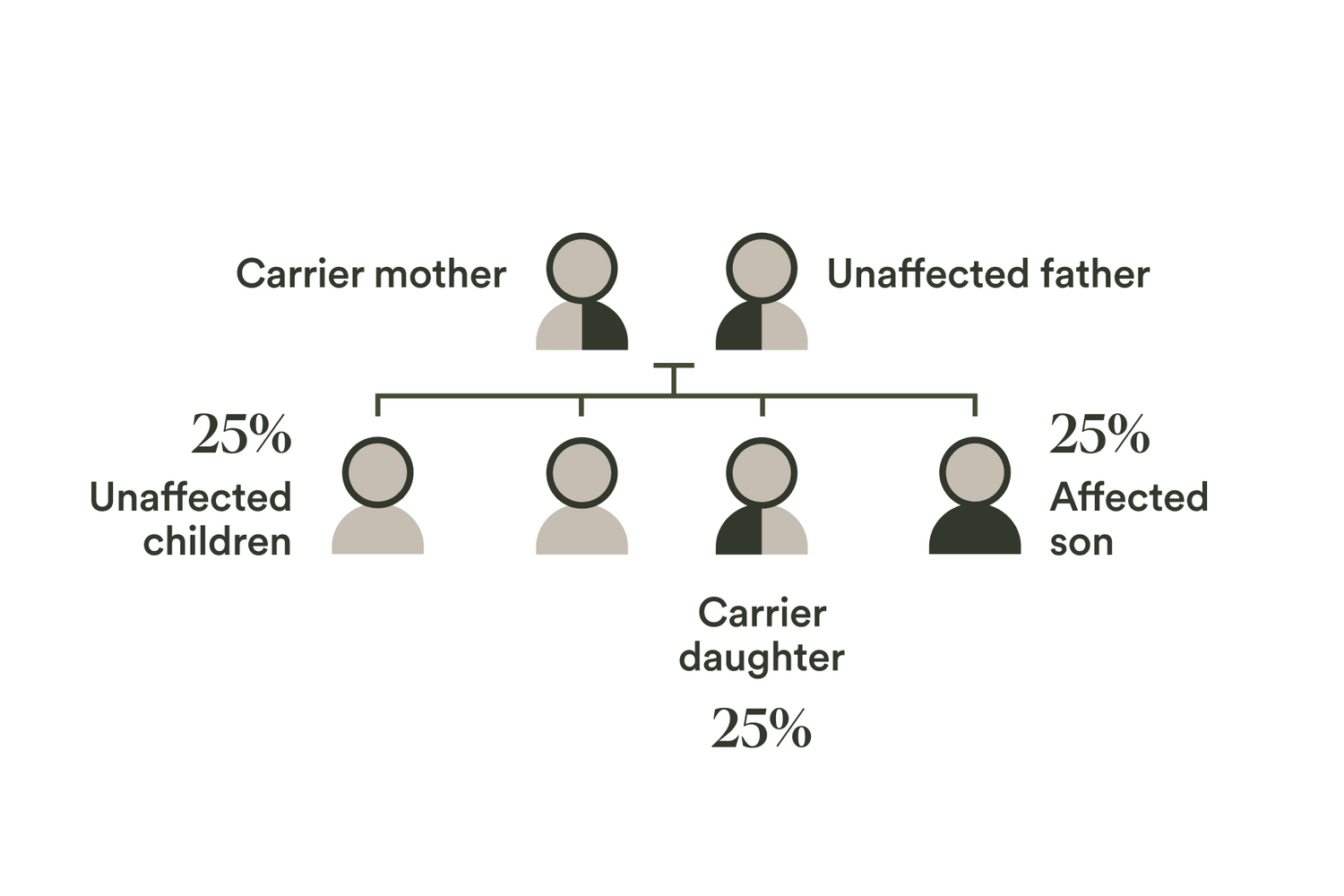
| AAAS | Achalasia-addisonianism-alacrimia syndrome | Autosomal recessive |
| ABCA12 | Congenital ichthyosis ABCA12-related | Autosomal recessive |
| ABCA3 | Surfactant metabolism dysfunction pulmonary 3 | Autosomal recessive |
| ABCA4 | Stargardt disease | Autosomal recessive |
| ABCB11 | Progressive familial intrahepatic cholestasis | Autosomal recessive |
| ABCB4 | Progressive familial intrahepatic cholestasis | Autosomal recessive |
| ABCC8 | Familial hyperinsulinism | Autosomal recessive |
| ABCD1 | Adrenoleukodystrophy X-linked | X-linked |
| ABCD4 | Methylmalonic aciduria and homocystinuria cblJ type | Autosomal recessive |
| ACAD9 | Acyl-CoA dehydrogenase-9 (ACAD9) deficiency | Autosomal recessive |
| ACADM | Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency | Autosomal recessive |
| ACADS | Short-chain acyl-coA dehydrogenase (SCAD) deficiency | Autosomal recessive |
| ACADSB | Short branched chain acyl-CoA dehydrogenase (SBCAD) deficiency | Autosomal recessive |
| ACADVL | Very long-chain acyl-CoA | Autosomal recessive |
| ACAT1 | 3-ketothiolase deficiency | Autosomal recessive |
| ACOX1 | Peroxisomal acyl-CoA oxidase deficiency | Autosomal recessive |
| ACSF3 | Combined malonic and methylmalonic aciduria | Autosomal recessive |
| ADA | Adenosine deaminase deficiency | Autosomal recessive |
| ADAMTS2 | Ehlers-Danlos syndrome dermatosparaxis type | Autosomal recessive |
| ADGRG1 | Bilateral frontoparietal polymicrogyria | Autosomal recessive |
| ADGRV1 | Usher syndrome type IIC | Autosomal recessive |
| ADK | Hypermethioninemia due to adenosine kinase deficiency | Autosomal recessive |
| AFF2 | Fragile XE syndrome | X-linked |
| AGA | Aspartylglucosaminuria | Autosomal recessive |
| AGL | Glycogen storage disease type III | Autosomal recessive |
| AGPAT2 | Congenital generalized lipodystrophy type 1 | Autosomal recessive |
| AGPS | Rhizomelic chondrodysplasia punctata type 3 | Autosomal recessive |
| AGXT | Primary hyperoxaluria type 1 | Autosomal recessive |
| AHCY | due to deficiency of S-adenosylhomocysteine hydrolase | Autosomal recessive |
| AHI1 | Joubert syndrome AHI1-related | Autosomal recessive |
| AIMP1 | Hypomyelinating leukodystrophy 3 | Autosomal recessive |
| AIPL1 | Childhood-onset severe retinal dystrophy AIPL1-related | Autosomal recessive |
| AIRE | Autoimmune polyendocrinopathy syndrome type I | Autosomal recessive |
| AK2 | Reticular dysgenesis | Autosomal recessive |
| AKR1D1 | Congenital Bile Acid Synthesis Defect 2 | Autosomal recessive |
| ALDH3A2 | Sjogren-Larsson syndrome | Autosomal recessive |
| ALDH4A1 | Hyperprolinemia type II | Autosomal recessive |
| ALDH7A1 | Pyridoxine-dependent epilepsy | Autosomal recessive |
| ALDOB | Hereditary fructose intolerance | Autosomal recessive |
| ALG1 | Congenital disorder of glycosylation type Ik | Autosomal recessive |
| ALG12 | Congenital disorder of glycosylation type Ig | Autosomal recessive |
| ALG3 | Congenital disorder of glycosylation type Id | Autosomal recessive |
| ALG6 | Congenital disorder of glycosylation type Ic | Autosomal recessive |
| ALMS1 | Alstrom syndrome | Autosomal recessive |
| ALOX12B | Autosomal recessive congenital ichthyosis 2 | Autosomal recessive |
| ALOXE3 | Congenital ichthyosiform erythroderma | Autosomal recessive |
| ALPL | Hypophosphatasia | Autosomal recessive |
| AMH | Persistent mullerian duct syndrome type I | Autosomal recessive |
| AMHR2 | Persistent mullerian duct syndrome type II | Autosomal recessive |
| AMN | Megaloblastic anemia 1 | Autosomal recessive |
| AMPD2 | Pontocerebellar hypoplasia type 9 | Autosomal recessive |
| AMT | Glycine encephalopathy | Autosomal recessive |
| ANO10 | Spinocerebellar ataxia 10 | Autosomal recessive |
| ANO5 | Limb girdle muscular dystrophy type 2L | Autosomal recessive |
| ANTXR2 | Hyaline fibromatosis syndrome | Autosomal recessive |
| AP1S1 | MEDNIK syndrome | Autosomal recessive |
| AP1S2 | X-linked Intellectual disability AP1S2-related | X-linked |
| AP3B1 | Hermansky-Pudlak syndrome 2 | Autosomal recessive |
| AP3D1 | Hermansky-Pudlak syndrome 10 | Autosomal recessive |
| APOPT1 | Mitochondrial complex IV deficiency | Autosomal recessive |
| AQP2 | Nephrogenic diabetes insipidus | Autosomal recessive |
| AR | Androgen insensitivity syndrome | X-linked |
| ARG1 | Arginase deficiency | Autosomal recessive |
| ARL13B | Joubert syndrome ARL13B-related | Autosomal recessive |
| ARL6 | ARL6-related disorders | Autosomal recessive |
| ARSA | Metachromatic leukodystrophy | Autosomal recessive |
| ARSB | Mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome) | Autosomal recessive |
| ARSE | Chondrodysplasia punctata type 1 X-linked | X-linked |
| ARX | X-linked intellectual disability ARX-related | X-linked |
| ASL | Argininosuccinate lyase deficiency | Autosomal recessive |
| ASNS | Asparagine synthetase deficiency | Autosomal recessive |
| ASPA | Canavan disease | Autosomal recessive |
| ASS1 | Citrullinemia | Autosomal recessive |
| ATM | Ataxia-telangiectasia | Autosomal recessive |
| ATP13A2 | Kufor-Rakeb syndrome | Autosomal recessive |
| ATP6V0A2 | Cutis laxa type IIA | Autosomal recessive |
| ATP6V0A4 | Renal tubular acidosis | Autosomal recessive |
| ATP6V1B1 | Renal tubular acidosis with deafness | Autosomal recessive |
| ATP6V1E1 | Cutis laxa type IIC | Autosomal recessive |
| ATP7A | Menkes disease | X-linked |
| ATP7B | Wilson disease | Autosomal recessive |
| ATP8B1 | Progressive familial intrahepatic cholestasis | Autosomal recessive |
| ATRX | Alpha thalassemia X-linked intellectual disability syndrome | X-linked |
| AVPR2 | Nephrogenic diabetes insipidus | X-linked |
| B9D1 | Joubert syndrome 27 | Autosomal recessive |
| B9D2 | Meckel syndrome 10 | Autosomal recessive |
| BBS1 | Bardet-Biedl syndrome type 1 | Autosomal recessive |
| BBS10 | Bardet-Biedl syndrome type 10 | Autosomal recessive |
| BBS12 | Bardet-Biedl syndrome type 12 | Autosomal recessive |
| BBS2 | BBS2-related ciliopathies | Autosomal recessive |
| BBS4 | Bardet-Biedl syndrome 4 | Autosomal recessive |
| BBS5 | Bardet-Biedl syndrome 5 | Autosomal recessive |
| BBS7 | Bardet-Biedl syndrome 7 | Autosomal recessive |
| BBS9 | Bardet-Biedl syndrome 9 | Autosomal recessive |
| BCHE | Butyrylcholinesterase deficiency | Autosomal recessive |
| BCKDHA | Maple syrup urine disease type Ia | Autosomal recessive |
| BCKDHB | Maple syrup urine disease type Ib | Autosomal recessive |
| BCS1L | Mitochondrial complex III deficiency | Autosomal recessive |
| BLM | Bloom syndrome | Autosomal recessive |
| BLOC1S3 | Hermansky-Pudlak syndrome 8 | Autosomal recessive |
| BLOC1S6 | Hermansky-Pudlak syndrome 9 | Autosomal recessive |
| BMP1 | Osteogenesis imperfecta type XIII | Autosomal recessive |
| BMPER | Diaphanospondylodysostosis | Autosomal recessive |
| BRIP1 | Fanconi anemia group J | Autosomal recessive |
| BRWD3 | X-linked intellectual disability BRWD3-related | X-linked |
| BSND | Bartter syndrome | Autosomal recessive |
| BTD | Biotinidase deficiency | Autosomal recessive |
| BTK | X-linked agammaglobulinemia | X-linked |
| C19orf12 | Mitochondrial membrane protein-associated neurodegeneration | Autosomal recessive |
| C8orf37 | Bardet-Biedl Syndrome 21 | Autosomal recessive |
| CAD | Early Infantile Epileptic Encephalopathy 50 | Autosomal recessive |
| CANT1 | Desbuquois dysplasia 1 | Autosomal recessive |
| CAPN3 | Limb-girdle muscular dystrophy type 2A | Autosomal recessive |
| CASP14 | Congenital Ichthyosis 12 | Autosomal recessive |
| CASQ2 | Catecholaminergic polymorphic ventricular tachycardia | Autosomal recessive |
| CASR | Neonatal hyperparathyroidism | Autosomal recessive |
| CAVIN1 | Congenital Generalized Lipodystrophy 4 | Autosomal recessive |
| CBS | Homocystinuria due to cystathionine | Autosomal recessive |
| CC2D1A | Autosomal recessive intellectual developmental disorder 3 | Autosomal recessive |
| CC2D2A | Joubert syndrome 9 | Autosomal recessive |
| CCDC103 | Primary ciliary dyskinesia type 17 | Autosomal recessive |
| CCDC151 | Primary ciliary dyskinesia type 30 | Autosomal recessive |
| CCDC39 | Primary ciliary dyskinesia type 14 | Autosomal recessive |
| CCDC8 | 3-M Syndrome | Autosomal recessive |
| CCDC88C | Congenital hydrocephalus 1 | Autosomal recessive |
| CD247 | Severe Combined Immunodeficiency | Autosomal recessive |
| CD3D | Severe Combined Immunodeficiency | Autosomal recessive |
| CD3E | Severe Combined Immunodeficiency | Autosomal recessive |
| CD3G | Severe Combined Immunodeficiency | Autosomal recessive |
| CD40LG | Hyper IgM syndrome X-linked | X-linked |
| CD59 | CD59 deficiency | Autosomal recessive |
| CD8A | Familial CD8 Deficiency | Autosomal recessive |
| CDAN1 | Dyserythropoietic congenital anemia type Ia | Autosomal recessive |
| CDCA7 | Immunodeficiency-centromeric instability-facial anomalies syndrome 3 | Autosomal recessive |
| CDH23 | Usher syndrome type 1D | Autosomal recessive |
| CEP104 | Joubert syndrome 25 | Autosomal recessive |
| CEP152 | CEP152-related disorders | Autosomal recessive |
| CEP290 | CEP290-related Ciliopathies | Autosomal recessive |
| CERKL | Retinitis pigmentosa 26 | Autosomal recessive |
| CERS3 | Congenital ichthyosis 9 | Autosomal recessive |
| CFTR | Cystic Fibrosis | Autosomal recessive |
| CHAT | Congenital myasthenic syndrome 6 | Autosomal recessive |
| CHM | Choroideremia | X-linked |
| CHMP1A | Pontocerebellar hypoplasia type 8 | Autosomal recessive |
| CHRNE | Congenital myasthenic syndrome | Autosomal recessive |
| CHRNG | Multiple pterygium syndrome | Autosomal recessive |
| CHST6 | Macular corneal dystrophy CHST6-related | Autosomal recessive |
| CIB2 | Nonsyndromic hearing loss 48 | Autosomal recessive |
| CIITA | Bare lymphocyte syndrome type II | Autosomal recessive |
| CLCF1 | Crisponi cold-induced sweating syndrome 2 | Autosomal recessive |
| CLCN1 | Autosomal recessive congenital myotonia | Autosomal recessive |
| CLCN5 | Dent disease | X-linked |
| CLCNKB | Bartter syndrome | Autosomal recessive |
| CLN3 | Neuronal ceroid lipofuscinosis | Autosomal recessive |
| CLN5 | Neuronal ceroid lipofuscinosis 5 | Autosomal recessive |
| CLN6 | Neuronal ceroid lipofuscinosis CLN6-related | Autosomal recessive |
| CLN8 | Neuronal ceroid lipofuscinosis CLN8-related | Autosomal recessive |
| CLP1 | Pontocerebellar hypoplasia type 10 | Autosomal recessive |
| CLRN1 | Usher syndrome type 3A | Autosomal recessive |
| CNGA1 | Retinitis Pigmentosa CNGA1-related | Autosomal recessive |
| CNGA3 | CNGA3-related retinopathy | Autosomal recessive |
| CNGB1 | Retinitis Pigmentosa CNGB1-related | Autosomal recessive |
| CNGB3 | Achromatopsia | Autosomal recessive |
| CNTNAP2 | Cortical dysplasia-focal epilepsy syndrome | Autosomal recessive |
| COASY | Pontocerebellar hypoplasia type 12 | Autosomal recessive |
| COL11A2 | COL11A2-related disorders | Autosomal recessive |
| COL17A1 | Junctional epidermolysis bullosa | Autosomal recessive |
| COL27A1 | Steel syndrome | Autosomal recessive |
| COL4A3 | Alport syndrome COL4A3-related | Autosomal recessive |
| COL4A4 | Alport syndrome COL4A4-related | Autosomal recessive |
| COL4A5 | Alport syndrome COL4A5-related | X-linked |
| COL7A1 | Dystrophic epidermolysis bullosa | Autosomal recessive |
| COLQ | Congenital myasthenic syndrome 5 | Autosomal recessive |
| COQ4 | Primary Coenzyme Q10 deficiency 7 | Autosomal recessive |
| CORO1A | Immunodeficiency 8 | Autosomal recessive |
| COX10 | Mitochondrial complex IV deficiency | Autosomal recessive |
| COX15 | Mitochondrial complex IV deficiency | Autosomal recessive |
| COX20 | Mitochondrial complex IV deficiency | Autosomal recessive |
| COX6B1 | Mitochondrial complex IV deficiency | Autosomal recessive |
| CP | Aceruloplasminemia | Autosomal recessive |
| CPLANE1 | Joubert syndrome 17 | Autosomal recessive |
| CPS1 | Carbamoylphosphate synthetase I deficiency | Autosomal recessive |
| CPT1A | Carnitine palmitoyltransferase IA deficiency | Autosomal recessive |
| CPT2 | Carnitine palmitoyltransferase II deficiency | Autosomal recessive |
| CRADD | Intellectual developmental disorder with variant lissencephaly | Autosomal recessive |
| CRB1 | CRB1-related retinopathy | Autosomal recessive |
| CRLF1 | Crisponi cold-induced sweating syndrome 1 | Autosomal recessive |
| CRTAP | Osteogenesis imperfecta type VII | Autosomal recessive |
| CRYL1 | GJB6-CRYL1 related nonsyndromic hearing loss | Autosomal recessive |
| CTC1 | Cerebroretinal microangiopathy with calcifications and cysts 1 | Autosomal recessive |
| CTNS | Cystinosis | Autosomal recessive |
| CTSA | Galactosialidosis | Autosomal recessive |
| CTSC | Papillon-Lefevre syndrome | Autosomal recessive |
| CTSD | Neuronal ceroid lipofuscinosis CTSD-related | Autosomal recessive |
| CTSF | Neuronal ceroid lipofuscinosis 13 | Autosomal recessive |
| CTSK | Pycnodysostosis | Autosomal recessive |
| CUL4B | X-linked intellectual disability CUL4B-related | X-linked |
| CUL7 | Three M syndrome 1 | Autosomal recessive |
| CWC27 | Retinitis pigmentosa with or without skeletal anomalies | Autosomal recessive |
| CYBA | Chronic granulomatous disease | Autosomal recessive |
| CYBB | Chronic granulomatous disease X-linked | X-linked |
| CYP11A1 | Congenital adrenal insufficiency | Autosomal recessive |
| CYP11B1 | Congenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency | Autosomal recessive |
| CYP11B2 | Corticosterone methyloxidase deficiency | Autosomal recessive |
| CYP17A1 | Congenital adrenal hyperplasia due to 17-alpha-hydroxylase deficiency | Autosomal recessive |
| CYP19A1 | Aromatase deficiency | Autosomal recessive |
| CYP1B1 | Primary congenital glaucoma | Autosomal recessive |
| CYP21A2 | Congenital adrenal hyperplasia due to 21-hydroxylase deficiency | Autosomal recessive |
| CYP27A1 | Cerebrotendinous xanthomatosis | Autosomal recessive |
| CYP27B1 | Vitamin D–dependent rickets type 1 | Autosomal recessive |
| CYP4F22 | Congenital ichthyosis 5 | Autosomal recessive |
| CYP7B1 | Congenital bile acid synthesis defect 3 | Autosomal recessive |
| DBT | Maple syrup urine disease type II | Autosomal recessive |
| DCAF17 | Woodhouse-Sakati syndrome | Autosomal recessive |
| DCLRE1C | Severe combined immunodeficiency with sensitivity to ionizing radiation | Autosomal recessive |
| DCX | Lissencephaly X-linked | X-linked |
| DDB2 | Xeroderma pigmentosum group E | Autosomal recessive |
| DDC | Aromatic l-amino acid decarboxylase deficiency | Autosomal recessive |
| DDR2 | Spondylometaepiphyseal dysplasia | Autosomal recessive |
| DDX11 | Warsaw breakage syndrome | Autosomal recessive |
| DGUOK | Mitochondrial DNA depletion syndrome 3 | Autosomal recessive |
| DHCR24 | Desmosterolosis | Autosomal recessive |
| DHCR7 | Smith-Lemli-Opitz syndrome | Autosomal recessive |
| DHDDS | Retinitis pigmentosa 59 | Autosomal recessive |
| DKC1 | X-linked dyskeratosis congenita | X-linked |
| DLAT | Pyruvate dehydrogenase E2 | Autosomal recessive |
| DLD | Dihydrolipoamide dehydrogenase deficiency | Autosomal recessive |
| DLG3 | X-linked intellectual disability DLG3-related | X-linked |
| DLL3 | Spondylocostal dysostosis 1 | Autosomal recessive |
| DMD | Dystrophinopathies | X-linked |
| DNAH5 | Primary ciliary dyskinesia DNAH5-related | Autosomal recessive |
| DNAI1 | Primary ciliary dyskinesia DNAI1-related | Autosomal recessive |
| DNAI2 | Primary ciliary dyskinesia DNAI2-related | Autosomal recessive |
| DNAL1 | Primary ciliary dyskinesia DNAL1-related | Autosomal recessive |
| DNMT3B | ICF Syndrome | Autosomal recessive |
| DOCK8 | Hyper-IgE syndrome due to DOCK8 defiency | Autosomal recessive |
| DOK7 | Congenital myasthenic syndrome DOK7-related | Autosomal recessive |
| DOLK | Congenital disorder of glycosylation type Im | Autosomal recessive |
| DPYD | Dihydropyrimidine dehydrogenase deficiency | Autosomal recessive |
| DTNBP1 | Hermansky-Pudlak syndrome 7 | Autosomal recessive |
| DUOX2 | Congenital hypothyroidism DUOX2-related | Autosomal recessive |
| DUOXA2 | Congenital hypothyroidism DUOXA2-related | Autosomal recessive |
| DYNC2H1 | Short-rib thoracic dysplasia 3 with or without polydactyly | Autosomal recessive |
| DYSF | Limb-girdle muscular dystrophy type 2B | Autosomal recessive |
| EDA | Hypohidrotic ectodermal dysplasia | X-linked |
| EFEMP2 | Cutis laxa type 1B | Autosomal recessive |
| EIF2AK3 | Wolcott-Rallison Syndrome | Autosomal recessive |
| EIF2B1 | Leukoencephalopathy with vanishing white matter | Autosomal recessive |
| EIF2B2 | Leukoencephalopathy with vanishing white matter | Autosomal recessive |
| EIF2B3 | Leukoencephalopathy with vanishing white matter | Autosomal recessive |
| EIF2B4 | Leukoencephalopathy with vanishing white matter | Autosomal recessive |
| EIF2B5 | Leukoencephalopathy with vanishing white matter | Autosomal recessive |
| ELP1 | Familial Dysautonomia | Autosomal recessive |
| EMD | Emery-Dreifuss muscular dystrophy | X-linked |
| EPB42 | Spherocytosis type 5 | Autosomal recessive |
| ERBB3 | Familial visceral neuropathy type 1 | Autosomal recessive |
| ERCC2 | ERCC2-related disorders | Autosomal recessive |
| ERCC3 | ERCC3-related photosensitivity | Autosomal recessive |
| ERCC4 | ERCC4-related disorders | Autosomal recessive |
| ERCC5 | Xeroderma Pigmentosa group G | Autosomal recessive |
| ERCC6 | ERCC6-related disorders | Autosomal recessive |
| ERCC8 | Cockayne syndrome type A | Autosomal recessive |
| ESCO2 | Roberts syndrome | Autosomal recessive |
| ETFA | Glutaric aciduria IIA | Autosomal recessive |
| ETFB | Glutaric aciduria IIB | Autosomal recessive |
| ETFDH | Glutaric aciduria IIC | Autosomal recessive |
| ETHE1 | Ethylmalonic encephalopathy | Autosomal recessive |
| EVC | EVC-related bone growth disorders | Autosomal recessive |
| EVC2 | EVC2-related bone growth disorders | Autosomal recessive |
| EXOSC3 | Pontocerebellar hypoplasia type 1B | Autosomal recessive |
| EYS | Retinitis pigmentosa 25 | Autosomal recessive |
| F11 | Factor XI deficiency | Autosomal recessive |
| F2 | Prothrombin-related conditions | Autosomal recessive |
| F5 | Factor V deficiency | Autosomal recessive |
| F7 | Factor VII deficiency | Autosomal recessive |
| F8 | Hemophilia A | X-linked |
| F9 | Hemophilia B | X-linked |
| FA2H | Spastic paraplegia type 35 | Autosomal recessive |
| FAH | Tyrosinemia type 1 | Autosomal recessive |
| FAM126A | Hypomyelinating leukodystropy type 5 | Autosomal recessive |
| FAM161A | Retinitis pigmentosa 28 | Autosomal recessive |
| FANCA | Fanconi anemia group A | Autosomal recessive |
| FANCB | Fanconi anemia group B | X-linked |
| FANCC | Fanconi anemia group C | Autosomal recessive |
| FANCD2 | Fanconi anemia group D2 | Autosomal recessive |
| FANCE | Fanconi anemia group E | Autosomal recessive |
| FANCF | Fanconi anemia group F | Autosomal recessive |
| FANCG | Fanconi anemia group G | Autosomal recessive |
| FANCI | Fanconi anemia group I | Autosomal recessive |
| FANCL | Fanconi anemia group L | Autosomal recessive |
| FBP1 | Fructose-1 6-bisphosphatase | Autosomal recessive |
| FBXL4 | Mitochondrial DNA depletion syndrome 13 | Autosomal recessive |
| FGD1 | X-linked Aarskog-Scott syndrome | X-linked |
| FH | Fumarase deficiency | Autosomal recessive |
| FHL1 | FHL1-related neuromuscular disorders | X-linked |
| FKBP10 | Osteogenesis imperfecta type XI | Autosomal recessive |
| FKRP | FKRP Alpha-dystroglycanopathies | Autosomal recessive |
| FKTN | FKTN Alpha-dystroglycanopathies | Autosomal recessive |
| FMO3 | Trimethylaminuria | Autosomal recessive |
| FMR1 | Fragile X syndrome | X-linked |
| FOLR1 | Cerebral folate deficiency | Autosomal recessive |
| FOXN1 | T-cell immunodeficiency with thymic aplasia | Autosomal recessive |
| FOXP3 | IPEX syndrome | X-linked |
| FOXRED1 | Mitochondrial complex I deficiency | Autosomal recessive |
| FRAS1 | Fraser syndrome | Autosomal recessive |
| FREM2 | Fraser syndrome | Autosomal recessive |
| FTCD | Glutamate formiminotransferase deficiency | Autosomal recessive |
| FTSJ1 | X-linked intellectual disability FTSJ1-related | X-linked |
| FUCA1 | Fucosidosis | Autosomal recessive |
| FXN | Friedreich ataxia | Autosomal recessive |
| G6PC | Glycogen storage disease type 1a | Autosomal recessive |
| G6PC3 | Severe congenital neutropenia 4 | Autosomal recessive |
| G6PD | Glucose-6-phosphate dehydrogenase deficiency | X-linked |
| GAA | Pompe disease | Autosomal recessive |
| GALC | Krabbe disease | Autosomal recessive |
| GALE | Galactose epimerase deficiency | Autosomal recessive |
| GALK1 | Galactokinase deficiency | Autosomal recessive |
| GALNS | Mucopolysaccharidosis IVA (Morquio syndrome A) | Autosomal recessive |
| GALNT3 | Familial hyperphosphatemic tumoral calcinosis | Autosomal recessive |
| GALT | Galactosemia | Autosomal recessive |
| GAMT | Guanidinoacetate methyltransferase deficiency | Autosomal recessive |
| GATM | Cerebral creatine deficiency syndrome 3 | Autosomal recessive |
| GBA | Gaucher disease | Autosomal recessive |
| GBE1 | Glycogen storage disease IV | Autosomal recessive |
| GCDH | Glutaric aciduria type I | Autosomal recessive |
| GDAP1 | Charcot-Marie-Tooth disease GDAP1-related | Autosomal recessive |
| GDF5 | Du Pan Syndrome | Autosomal recessive |
| GFM1 | Combined oxidative phosphorylation deficiency GFM1-related | Autosomal recessive |
| GFPT1 | Congenital myasthenic syndrome 12 | Autosomal recessive |
| GHR | Growth hormone insensitivity syndrome | Autosomal recessive |
| GHRHR | Isolated growth hormone deficiency | Autosomal recessive |
| GJB1 | Charcot-Marie-Tooth disease X-linked type 1 | X-linked |
| GJB2 | Nonsyndromic hearing loss 1A | Autosomal recessive |
| GJB6 | GJB6-CRYL1 related nonsyndromic hearing loss | Autosomal recessive |
| GLA | Fabry disease | X-linked |
| GLB1 | GLB1-related gangliosidoses | Autosomal recessive |
| GLDC | Glycine encephalopathy GLDC-related | Autosomal recessive |
| GLE1 | Lethal congenital contracture syndrome 1 | Autosomal recessive |
| GNE | Inclusion body myopathy type 2 (Nonaka myopathy) | Autosomal recessive |
| GNPAT | Rhizomelic chondrodysplasia punctata type 2 | Autosomal recessive |
| GNPTAB | Mucolipidosis II & III | Autosomal recessive |
| GNPTG | Mucolipidosis III gamma | Autosomal recessive |
| GNRHR | Hypogonadotropic hypogonadism GNRHR-related | Autosomal recessive |
| GNS | Mucopolysaccharidosis IIID (Sanfilippo syndrome D) | Autosomal recessive |
| GORAB | Geroderma osteodysplasticum | Autosomal recessive |
| GP1BA | Bernard-Soulier syndrome type A1 | Autosomal recessive |
| GP9 | Bernard-Soulier syndrome type C | Autosomal recessive |
| GPR143 | X-linked Ocular albinism GPR143-related | X-linked |
| GRHPR | Primary hyperoxaluria type II | Autosomal recessive |
| GRIP1 | Fraser syndrome | Autosomal recessive |
| GSS | Glutathione synthetase deficiency | Autosomal recessive |
| GUCY2D | Leber congenital amaurosis 1 | Autosomal recessive |
| GUSB | Mucopolysaccharidosis type VII | Autosomal recessive |
| GYS2 | Glycogen storage disease type 0 liver | Autosomal recessive |
| HADH | Familial hyperinsulinemic hypoglycemia 4 | Autosomal recessive |
| HADHA | Trifunctional protein deficiency | Autosomal recessive |
| HADHB | Trifunctional protein deficiency | Autosomal recessive |
| HAMP | Hemochromatosis type 2B | Autosomal recessive |
| HAX1 | Severe congenital neutropenia HAX1-related | Autosomal recessive |
| HBA1 | Alpha thalassemia | Autosomal recessive |
| HBA2 | Alpha thalassemia | Autosomal recessive |
| HBB | Sickle cell disease Hemoglobin C disease Beta thalassemia | Autosomal recessive |
| HCFC1 | Methylmalonic acidemia with homocystinuria type cblX | X-linked |
| HELLS | Immunodeficiency Centromeric region instability Facial anomalies syndrome | Autosomal recessive |
| HEXA | Tay-Sachs disease | Autosomal recessive |
| HEXB | Sandhoff disease | Autosomal recessive |
| HFE | Hereditary Hemochromatosis | Autosomal recessive |
| HGD | Alkaptonuria | Autosomal recessive |
| HGSNAT | Mucopolysaccharidosis type IIIC (Sanfilippo syndrome C) | Autosomal recessive |
| HINT1 | Neuromyotonia and axonal neuropathy | Autosomal recessive |
| HJV | Hemochromatosis type 2A | Autosomal recessive |
| HLCS | Holocarboxylase synthetase deficiency | Autosomal recessive |
| HMGCL | 3-hydroxy-3-methylglutaryl-CoA lyase deficiency | Autosomal recessive |
| HMGCS2 | 3-hydroxy-3-methylglutaryl-CoA synthase 2 deficiency | Autosomal recessive |
| HOGA1 | Primary hyperoxaluria type III | Autosomal recessive |
| HPD | Tyrosinemia type III | Autosomal recessive |
| HPS1 | Hermansky-Pudlak syndrome 1 | Autosomal recessive |
| HPS3 | Hermansky-Pudlak syndrome 3 | Autosomal recessive |
| HPS4 | Hermansky-Pudlak syndrome 4 | Autosomal recessive |
| HPS5 | Hermansky-Pudlak syndrome 5 | Autosomal recessive |
| HPS6 | Hermansky-Pudlak syndrome 6 | Autosomal recessive |
| HSD17B10 | HSD10 mitochondrial disease | X-linked |
| HSD17B3 | 17-Beta-Hydroxysteroid Dehydrogenase Deficiency | Autosomal recessive |
| HSD17B4 | D-bifunctional protein deficiency | Autosomal recessive |
| HSD3B2 | Congenital adrenal hyperplasia due to 3-beta-hydroxysteroid dehydrogenase 2 deficiency | Autosomal recessive |
| HSD3B7 | Congenital bile acid synthesis defect 1 | Autosomal recessive |
| HYAL1 | Mucopolysaccharidosis type IX | Autosomal recessive |
| HYLS1 | Hydrolethalus syndrome | Autosomal recessive |
| IDH3B | Retinitis pigmentosa IDH3B-related | Autosomal recessive |
| IDS | Mucopolysaccharidosis type II (Hunter syndrome) | X-linked |
| IDUA | Mucopolysaccharidosis type I (Hurler syndrome) | Autosomal recessive |
| IFT140 | IFT140-related disorders | Autosomal recessive |
| IGHMBP2 | IGHMBP2-related neuropathies | Autosomal recessive |
| IGSF1 | X-linked central hypothyroidism and testicular enlargement | X-linked |
| IKBKB | Immunodeficiency 15B | Autosomal recessive |
| IL1RAPL1 | X-linked intellectual disability IL1RAPL1-related | X-linked |
| IL2RA | Immunodeficiency due to CD25 deficiency | Autosomal recessive |
| IL2RG | X-linked severe combined immunodeficiency | X-linked |
| IL7R | Severe Combined Immunodeficiency 104 | Autosomal recessive |
| INPP5E | Joubert syndrome 1 | Autosomal recessive |
| INVS | Nephronophthisis 2 | Autosomal recessive |
| ITGA2B | Glanzmann thrombasthenia | Autosomal recessive |
| ITGA6 | Junctional epidermolysis bullosa | Autosomal recessive |
| ITGB3 | Glanzmann thrombasthenia | Autosomal recessive |
| ITGB4 | Junctional epidermolysis bullosa | Autosomal recessive |
| ITPA | Developmental and epileptic encephalopathy 35 | Autosomal recessive |
| IVD | Isovaleric Acidemia | Autosomal recessive |
| IYD | Thyroid dyshormonogenesis IYD-related | Autosomal recessive |
| JAK3 | Severe combined immunodeficiency JAK3-related | Autosomal recessive |
| KCNJ1 | Bartter syndrome | Autosomal recessive |
| KCNJ11 | KCNJ11-related hyperinsulinism | Autosomal recessive |
| KCTD7 | Progressive myoclonic epilepsy type 3 | Autosomal recessive |
| KDM5C | X-linked intellectual disability KDM5C-related | X-linked |
| KIF14 | Primary Autosomal Recessive Microcephaly 20 | Autosomal recessive |
| L1CAM | L1 syndrome | X-linked |
| LAMA2 | Muscular dystrophy LAMA2-related | Autosomal recessive |
| LAMA3 | Junctional epidermolysis bullosa 2 | Autosomal recessive |
| LAMB3 | Junctional epidermolysis bullosa LAMB3-related | Autosomal recessive |
| LAMC2 | Junctional epidermolysis bullosa LAMC2-related | Autosomal recessive |
| LARS | Infantile liver failure syndrome 1 | Autosomal recessive |
| LCA5 | Leber congenital amaurosis 5 | Autosomal recessive |
| LCK | Immunodeficiency 22 | Autosomal recessive |
| LDLR | Familial Hypercholesterolemia | Autosomal recessive |
| LDLRAP1 | Familial Hypercholesterolemia | Autosomal recessive |
| LHCGR | Leydig cell hypoplasia | Autosomal recessive |
| LHX3 | Combined pituitary hormone deficiency 3 | Autosomal recessive |
| LIFR | Stuve-Wiedemann syndrome | Autosomal recessive |
| LIG4 | LIG4 syndrome | Autosomal recessive |
| LIPA | Lysosomal acid lipase deficiency | Autosomal recessive |
| LIPN | Congenital Ichthyosis 8 | Autosomal recessive |
| LMAN1 | Combined factor V and VIII deficiency | Autosomal recessive |
| LMBRD1 | Methylmalonic aciduria and homocystinuria cblF type | Autosomal recessive |
| LOXHD1 | Nonsyndromic hearing loss 77 | Autosomal recessive |
| LPAR6 | Hypotrichosis 8 | Autosomal recessive |
| LPL | Familial lipoprotein lipase deficiency | Autosomal recessive |
| LRAT | Leber congenital amaurosis 14 | Autosomal recessive |
| LRP2 | Donnai–Barrow syndrome | Autosomal recessive |
| LRPPRC | Leigh syndrome with Complex IV deficiency | Autosomal recessive |
| LTBP4 | Cutis laxa with severe pulmonary gastrointestinal and urinary abnormalities | Autosomal recessive |
| LYST | Chediak-Higashi syndrome | Autosomal recessive |
| MAK | Retinitis Pigmentosa 62 | Autosomal recessive |
| MALT1 | Immunodeficiency 12 | Autosomal recessive |
| MAN2B1 | Alpha-Mannosidosis | Autosomal recessive |
| MANBA | Beta-Mannosidosis | Autosomal recessive |
| MAT1A | Methionine adenosyltransferase deficiency | Autosomal recessive |
| MCCC1 | 3-Methylcrotonyl-CoA carboxylase 1 deficiency (3-MCC deficiency) | Autosomal recessive |
| MCCC2 | 3-Methylcrotonyl-CoA carboxylase 2 deficiency (3-MCC deficiency) | Autosomal recessive |
| MCEE | Methylmalonyl-CoA epimerase deficiency | Autosomal recessive |
| MCOLN1 | Mucolipidosis IV | Autosomal recessive |
| MCPH1 | Primary microcephaly 1 recessive | Autosomal recessive |
| MED17 | Postnatal Progressive Microcephaly with Seizures and Brain Atrophy | Autosomal recessive |
| MEFV | Familial Mediterranean fever | Autosomal recessive |
| MEGF8 | Carpenter syndrome 2 | Autosomal recessive |
| MESP2 | Spondylocostal dysostosis | Autosomal recessive |
| MFSD8 | Neuronal ceroid lipofuscinosis MFSD8-related | Autosomal recessive |
| MID1 | Opitz GBBB syndrome type I | X-linked |
| MKKS | Bardet-Biedl syndrome 6 | Autosomal recessive |
| MKS1 | MKS1-related ciliopathies | Autosomal recessive |
| MLC1 | Megalencephalic leukoencephalopathy with subcortical cysts | Autosomal recessive |
| MLYCD | Malonyl-CoA decarboxylase deficiency | Autosomal recessive |
| MMAA | Methylmalonic aciduria cblA type | Autosomal recessive |
| MMAB | Methylmalonic aciduria cblB type | Autosomal recessive |
| MMACHC | Methylmalonic aciduria and homocystinuria cblC type | Autosomal recessive |
| MMADHC | Methylmalonic aciduria and homocystinuria cblD type | Autosomal recessive |
| MPI | Congenital disorder of glycosylation | Autosomal recessive |
| MPL | Congenital amegakaryocytic thrombocytopenia | Autosomal recessive |
| MPV17 | Hepatocerebral mitochondrial DNA depletion syndrome MPV17-related | Autosomal recessive |
| MRE11 | Ataxia-Telangiectasia-Like Disorder 1 | Autosomal recessive |
| MTHFD1 | Combined immunodeficiency and megaloblastic anemia | Autosomal recessive |
| MTHFR | Homocystinuria MTHFR-related | Autosomal recessive |
| MTM1 | Myotubular myopathy X-linked | X-linked |
| MTMR2 | Charcot-Marie-Tooth disease type 4B1 | Autosomal recessive |
| MTR | Methylcobalamin deficiency type cblG | Autosomal recessive |
| MTRR | Homocystinuria-megaloblastic anemia cobalamin E type | Autosomal recessive |
| MTTP | Abetalipoproteinemia | Autosomal recessive |
| MUT | methylmalonyl–CoA mutase deficiency | Autosomal recessive |
| MVK | Mevalonate kinase deficiency | Autosomal recessive |
| MYO15A | Nonsyndromic hearing loss MYO15A-related | Autosomal recessive |
| MYO7A | MYO7A-related disorders | Autosomal recessive |
| NAGA | Schindler disease types 1 and 3 | Autosomal recessive |
| NAGLU | Mucopolysaccharidosis type IIIB (Sanfilippo syndrome B) | Autosomal recessive |
| NAGS | N-acetylglutamate synthase deficiency | Autosomal recessive |
| NBAS | SOPH syndrome | Autosomal recessive |
| NBEAL2 | Gray platelet syndrome | Autosomal recessive |
| NBN | Nijmegen breakage syndrome | Autosomal recessive |
| NCF2 | Chronic granulomatous disease 2 | Autosomal recessive |
| NCF4 | Chronic granulomatous disease 4 | Autosomal recessive |
| NDP | Norrie disease | X-linked |
| NDRG1 | Charcot-Marie-Tooth disease type 4D | Autosomal recessive |
| NDUFA11 | Mitochondrial complex I deficiency | Autosomal recessive |
| NDUFAF2 | Mitochondrial complex I deficiency | Autosomal recessive |
| NDUFAF5 | Mitochondrial complex I deficiency (Leigh syndrome) | Autosomal recessive |
| NDUFS4 | Mitochondrial complex I deficiency | Autosomal recessive |
| NDUFS6 | Mitochondrial complex I deficiency (Leigh syndrome) | Autosomal recessive |
| NDUFS7 | Mitochondrial complex I deficiency | Autosomal recessive |
| NDUFV1 | Mitochondrial complex I deficiency nuclear type 4 | Autosomal recessive |
| NEB | Nemaline myopathy | Autosomal recessive |
| NEU1 | Sialidosis type I and II | Autosomal recessive |
| NGLY1 | Congenital disorder of deglycosylation | Autosomal recessive |
| NHEJ1 | Severe combined immunodeficiency with microcephaly growth retardation and sensitivity to ionizing radiation | Autosomal recessive |
| NIPAL4 | Autosomal Recessive Congenital Ichthyosis 6 | Autosomal recessive |
| NONO | X-linked intellectual disability syndrome 34 | X-linked |
| NPC1 | Niemann-Pick disease type C1 | Autosomal recessive |
| NPC2 | Niemann-Pick disease type C2 | Autosomal recessive |
| NPHP1 | NPHP1-related ciliopathies | Autosomal recessive |
| NPHP3 | NPHP3-related ciliopathies | Autosomal recessive |
| NPHS1 | Congenital nephrotic syndrome type 1 1 | Autosomal recessive |
| NPHS2 | Congenital nephrotic syndrome type 2 | Autosomal recessive |
| NR0B1 | Congenital adrenal hypoplasia X-linked | X-linked |
| NR2E3 | NR2E3-related retinal dystrophies | Autosomal recessive |
| NTRK1 | Congenital insensitivity to pain with anhidrosis | Autosomal recessive |
| OAT | Gyrate atrophy of choroid and retina | Autosomal recessive |
| OBSL1 | 3M syndrome 2 | Autosomal recessive |
| OCA2 | Oculocutaneous albinism type II | Autosomal recessive |
| OCRL | OCRL-related disoders | X-linked |
| OPA3 | Costeff syndrome | Autosomal recessive |
| OPHN1 | X-linked intellectual disability- cerebellar hypoplasia syndrome | X-linked |
| OSTM1 | Osteopetrosis 5 | Autosomal recessive |
| OTC | Ornithine transcarbamylase deficiency | X-linked |
| OTOF | Nonsyndromic hearing loss OTOF-related | Autosomal recessive |
| P3H1 | Osteogenesis imperfecta type VIII | Autosomal recessive |
| PAH | Phenylalanine Hydroxylase deficiency (Phenylketonuria) | Autosomal recessive |
| PAK3 | X-linked intellectual disability PAK3- related | X-linked |
| PANK2 | Pantothenate kinase-associated neurodegeneration | Autosomal recessive |
| PC | Pyruvate carboxylase deficiency | Autosomal recessive |
| PCBD1 | Tetrahydrobiopterin deficiency PCBD1-related | Autosomal recessive |
| PCCA | Propionic acidemia PCCA-related | Autosomal recessive |
| PCCB | Propionic acidemia PCCB-related | Autosomal recessive |
| PCDH15 | PCDH15-related sensory loss | Autosomal recessive |
| PCNT | Microcephalic osteodysplastic primordial dwarfism type II | Autosomal recessive |
| PDE6A | Retinitis pigmentosa PDE6A-related | Autosomal recessive |
| PDHA1 | Pyruvate dehydrogenase E1-alpha deficiency | X-linked |
| PDHB | Pyruvate dehydrogenase E1-beta deficiency | Autosomal recessive |
| PDHX | Pyruvate dehydrogenase E3-binding protein deficiency | Autosomal recessive |
| PDP1 | Pyruvate dehydrogenase phosphatase deficiency | Autosomal recessive |
| PEPD | Prolidase deficiency | Autosomal recessive |
| PET100 | Mitochondrial complex IV deficiency | Autosomal recessive |
| PEX1 | Zellweger syndrome PEX1-related | Autosomal recessive |
| PEX10 | Zellweger syndrome PEX10-related | Autosomal recessive |
| PEX11B | Zellweger spectrum disorder | Autosomal recessive |
| PEX12 | Zellweger syndrome PEX12-related | Autosomal recessive |
| PEX13 | Zellweger spectrum disorder | Autosomal recessive |
| PEX14 | Zellweger spectrum disorder | Autosomal recessive |
| PEX16 | Zellweger spectrum disorder | Autosomal recessive |
| PEX19 | Zellweger spectrum disorder | Autosomal recessive |
| PEX2 | Zellweger syndrome PEX2-related | Autosomal recessive |
| PEX26 | Zellweger syndrome | Autosomal recessive |
| PEX3 | Zellweger spectrum disorder | Autosomal recessive |
| PEX5 | Zellweger spectrum disorder | Autosomal recessive |
| PEX6 | Zellweger syndrome PEX6-related | Autosomal recessive |
| PEX7 | Rhizomelic chondrodysplasia punctata type 1 | Autosomal recessive |
| PFKM | Glycogen storage disease VII | Autosomal recessive |
| PGK1 | Phosphoglycerate kinase 1 deficiency | X-linked |
| PGM3 | Immunodeficiency 23 | Autosomal recessive |
| PHF8 | X-linked intellectual disability Siderius type | X-linked |
| PHGDH | Phosphoglycerate dehydrogenase deficiency | Autosomal recessive |
| PHKA1 | Glycogen storage disease type IXd | X-linked |
| PHKA2 | Glycogen storage disease type IXa | X-linked |
| PHKB | Glycogen storage disease type IXb | Autosomal recessive |
| PHKG2 | Glycogen storage disease type IXc | Autosomal recessive |
| PHYH | Refsum disease | Autosomal recessive |
| PIGN | Multiple congenital anomalies hypotonia seizures syndrome 1 | Autosomal recessive |
| PIP5K1C | Lethal congenital contractural syndrome 3 | Autosomal recessive |
| PJVK | Nonsyndromic hearing loss 59 | Autosomal recessive |
| PKHD1 | Polycystic kidney disease PKHD1-related | Autosomal recessive |
| PLA2G6 | Infantile neuroaxonal dystrophy | Autosomal recessive |
| PLEKHG5 | PLEKHG5-related motor neuropathies | Autosomal recessive |
| PLOD1 | Ehlers-Danlos syndrome with kyphoscoliosis PLOD1-related | Autosomal recessive |
| PLOD2 | Bruck syndrome 2 | Autosomal recessive |
| PLP1 | PLP1-related disorders | X-linked |
| PMM2 | Congenital disorder of glycosylation type 1a | Autosomal recessive |
| PNP | Purine nucleoside phosphorylase deficiency | Autosomal recessive |
| PNPLA1 | Autosomal recessive congenital ichthyosis 10 | Autosomal recessive |
| PNPO | Pyridoxamine 5’-phosphate oxidase deficiency | Autosomal recessive |
| POC1A | Short stature onychodysplasia facial dysmorphism and hypotrichosis syndrome | Autosomal recessive |
| POLG | POLG-related disorders | Autosomal recessive |
| POLH | Xeroderma pigmentosum | Autosomal recessive |
| POLR1C | POLR1C-related disorders | Autosomal recessive |
| POMGNT1 | POMGNT1 Alpha-dystroglycanopathies | Autosomal recessive |
| POMT1 | POMT1 Alpha-dystroglycanopathies | Autosomal recessive |
| POMT2 | POMT2 Alpha-dystroglycanopathies | Autosomal recessive |
| POR | Antley-Bixler syndrome | Autosomal recessive |
| POU1F1 | Combined pituitary hormone deficiency | Autosomal recessive |
| POU3F4 | X-linked hearing loss POU3F4- related | X-linked |
| PPIB | Osteogenesis imperfecta type IX | Autosomal recessive |
| PPT1 | Neuronal ceroid lipofuscinosis PPT1-related | Autosomal recessive |
| PQBP1 | Renpenning syndrome | X-linked |
| PRCD | Retinitis pigmentosa 36 | Autosomal recessive |
| PRDM5 | Brittle cornea syndrome 2 | Autosomal recessive |
| PRF1 | Hemophagocytic lymphohistiocytosis familial 2 | Autosomal recessive |
| PRICKLE1 | Progressive myoclonic epilepsy type 1B | Autosomal recessive |
| PRKDC | PRKDC-related immunodeficiency | Autosomal recessive |
| PROP1 | Combined pituitary hormone deficiency 2 | Autosomal recessive |
| PRPS1 | PRPS1-related disorders | X-linked |
| PSAP | Metachromatic leukodystrophy due to saposin-b deficiency | Autosomal recessive |
| PTPRC | PTPRC related-severe combined immunodeficiency | Autosomal recessive |
| PTS | Tetrahydrobiopterin deficiency | Autosomal recessive |
| PUS1 | Mitochondrial myopathy and sideroblastic anemia 1 | Autosomal recessive |
| PYCR1 | Cutis laxa type IIB and type IIIB | Autosomal recessive |
| PYGL | Glycogen storage disease VI | Autosomal recessive |
| PYGM | Glycogen storage disease type V | Autosomal recessive |
| QDPR | Tetrahydrobiopterin deficiency QDPR-related | Autosomal recessive |
| RAB23 | Carpenter syndrome | Autosomal recessive |
| RAG1 | Omenn syndrome RAG1-related | Autosomal recessive |
| RAG2 | Omenn syndrome RAG2-related | Autosomal recessive |
| RAPSN | RAPSN-associated acetylcholine receptor deficiency | Autosomal recessive |
| RARS2 | Pontocerebellar hypoplasia type 6 | Autosomal recessive |
| RAX | Microphthalmia isolated 3 | Autosomal recessive |
| RD3 | Leber congenital amaurosis 12 | Autosomal recessive |
| RDH12 | Leber congenital amaurosis type 13 | Autosomal recessive |
| RDH5 | Fundus albipunctatus | Autosomal recessive |
| RFX5 | Bare lymphocyte syndrome type II | Autosomal recessive |
| RFXANK | MHC class II deficiency | Autosomal recessive |
| RFXAP | Bare lymphocyte syndrome type II | Autosomal recessive |
| RHAG | Rh Deficiency syndrome | Autosomal recessive |
| RLBP1 | Retinal dystrophy RLBP1-related | Autosomal recessive |
| RMRP | Cartilage-Hair Hypoplasia Anauxetic Dysplasia Spectrum Disorder | Autosomal recessive |
| RNASEH2A | Aicardi-Goutieres syndrome 4 | Autosomal recessive |
| RNASEH2B | Aicardi Goutieres syndrome 2 | Autosomal recessive |
| RNASEH2C | Aicardi-Goutieres syndrome 3 | Autosomal recessive |
| ROGDI | Kohlschutter-Tonz syndrome | Autosomal recessive |
| RP2 | X-linked Retinitis pigmentosa RP2-related | X-linked |
| RPE65 | RPE65-related retinopathy | Autosomal recessive |
| RPGR | X-linked Retinitis pigmentosa RPGR-related | X-linked |
| RPGRIP1 | Leber congenital amaurosis and Cone-rod dystrophy | Autosomal recessive |
| RPGRIP1L | RPGRIP1L-related ciliopathies | Autosomal recessive |
| RS1 | Juvenile retinoschisis X-linked | X-linked |
| RSPH9 | Primary ciliary dyskinesia 12 | Autosomal recessive |
| RTEL1 | Dyskeratosis congenita type 5 | Autosomal recessive |
| SACS | Autosomal recessive spastic ataxia of Charlevoix-Saguenay | Autosomal recessive |
| SAG | Retinitis pigmentosa 47 | Autosomal recessive |
| SAMD9 | Normophosphatemic Familial Tumoral Calcinosis | Autosomal recessive |
| SAMHD1 | Aicardi-Goutieres syndrome | Autosomal recessive |
| SARS2 | Hyperuricemia pulmonary hypertension renal failure and alkalosis syndrome | Autosomal recessive |
| SBDS | Shwachman-Diamond syndrome | Autosomal recessive |
| SCO1 | Mitochondrial complex IV deficiency | Autosomal recessive |
| SCO2 | Mitochondrial complex IV deficiency | Autosomal recessive |
| SDCCAG8 | Bardet-Biedl syndrome and Senior-Loken syndrome | Autosomal recessive |
| SDR9C7 | Autosomal recessive congenital ichthyosis | Autosomal recessive |
| SEC23B | Congenital dyserythropoietic anemia type II | Autosomal recessive |
| SELENON | Rigid spine muscular dystrophy | Autosomal recessive |
| SEPSECS | Pontocerebellar hypoplasia type 2D | Autosomal recessive |
| SERPINA1 | Alpha-1 antitrypsin deficiency | Autosomal recessive |
| SERPINF1 | Osteogenesis imperfecta type VI | Autosomal recessive |
| SGCA | Limb-girdle muscular dystrophy type 2D | Autosomal recessive |
| SGCB | Limb-girdle muscular dystrophy type 2E | Autosomal recessive |
| SGCD | Limb-girdle muscular dystrophy type 2F | Autosomal recessive |
| SGCG | Limb-girdle muscular dystrophy type 2C | Autosomal recessive |
| SGSH | Mucopolysaccharidosis IIIA (Sanfilippo syndrome A) | Autosomal recessive |
| SH3TC2 | Charcot-Marie-Tooth disease SH3TC2-related | Autosomal recessive |
| SKIV2L | Trichohepatoenteric syndrome 2 | Autosomal recessive |
| SLC12A1 | Bartter syndrome | Autosomal recessive |
| SLC12A3 | Gitelman syndrome | Autosomal recessive |
| SLC12A6 | Andermann syndrome | Autosomal recessive |
| SLC16A2 | Allan-Herndon-Dudley syndrome | X-linked |
| SLC17A5 | Sialic acid storage disorder | Autosomal recessive |
| SLC19A2 | Thiamine-responsive megaloblastic anemia syndrome | Autosomal recessive |
| SLC19A3 | Biotin-responsive basal ganglia disease | Autosomal recessive |
| SLC1A4 | Spastic tetraplegia thin corpus callosum and progressive microcephaly syndrome | Autosomal recessive |
| SLC22A5 | Systemic primary carnitine deficiency | Autosomal recessive |
| SLC25A13 | Citrin deficiency | Autosomal recessive |
| SLC25A15 | Hyperornithinemia-hyperammonemia-homocitrullinemia syndrome (Triple H syndrome) | Autosomal recessive |
| SLC25A20 | Carnitine-acylcarnitine translocase deficiency | Autosomal recessive |
| SLC26A2 | SLC26A2-related disorders | Autosomal recessive |
| SLC26A3 | Congenital secretory chloride diarrhea | Autosomal recessive |
| SLC26A4 | Pendred syndrome | Autosomal recessive |
| SLC27A4 | Ichthyosis prematurity syndrome | Autosomal recessive |
| SLC2A10 | Arterial tortuosity syndrome | Autosomal recessive |
| SLC2A2 | Fanconi-Bickel syndrome | Autosomal recessive |
| SLC34A3 | Hereditary hypophosphatemic rickets with hypercalciuria | Autosomal recessive |
| SLC35A3 | Arthrogryposis intellectual disability and seizures | Autosomal recessive |
| SLC37A4 | Glycogen storage disease type Ib | Autosomal recessive |
| SLC39A4 | Acrodermatitis enteropathica | Autosomal recessive |
| SLC3A1 | Cystinuria type I | Autosomal recessive |
| SLC45A2 | Oculocutaneous albinism type IV | Autosomal recessive |
| SLC46A1 | Hereditary folate malabsorption | Autosomal recessive |
| SLC4A1 | Distal Renal Tubular Acidosis | Autosomal recessive |
| SLC4A11 | Corneal endothelial dystrophy | Autosomal recessive |
| SLC5A5 | Thyroid dyshormonogenesis SLC5A5-related | Autosomal recessive |
| SLC6A19 | Hartnup disorder | Autosomal recessive |
| SLC6A8 | Creatine deficiency syndrome | X-linked |
| SLC7A7 | Lysinuric protein intolerance | Autosomal recessive |
| SLC7A9 | Cystinuria non-type I | Autosomal recessive |
| SMARCAL1 | Schimke immunoosseous dysplasia | Autosomal recessive |
| SMN1 | Spinal muscular atrophy | Autosomal recessive |
| SMPD1 | Niemann-Pick disease type A/B | Autosomal recessive |
| SNAP29 | Cerebral dysgenesis neuropathy ichthyosis and palmoplantar keratoderma syndrome | Autosomal recessive |
| SNX10 | Osteopetrosis 8 | Autosomal recessive |
| SP110 | Hepatic venoocclusive disease with immunodeficiency | Autosomal recessive |
| SPATA7 | Leber congenital amaurosis (LCA) and juvenile retinitis pigmentosa (RP) | Autosomal recessive |
| SPG11 | SPG11-related Neuromuscular Disorders | Autosomal recessive |
| SPG21 | Mast syndrome | Autosomal recessive |
| SPG7 | Spastic paraplegia type 7 | Autosomal recessive |
| SPINK5 | Netherton syndrome | Autosomal recessive |
| SPR | Sepiapterin Reductase Deficiency | Autosomal recessive |
| SRD5A2 | 5-alpha reductase deficiency | Autosomal recessive |
| ST3GAL5 | Salt and pepper developmental regression syndrome | Autosomal recessive |
| STAR | Lipoid congenital adrenal hyperplasia | Autosomal recessive |
| STK4 | Combined immunodeficiency due to STK4 deficiency | Autosomal recessive |
| STX11 | Familial hemophagocytic lymphohistiocytosis | Autosomal recessive |
| STXBP2 | Familial hemophagocytic lymphohistiocytosis | Autosomal recessive |
| SUCLA2 | Mitochondrial DNA depletion syndrome 5 | Autosomal recessive |
| SUMF1 | Multiple sulfatase deficiency | Autosomal recessive |
| SUOX | Sulfite oxidase deficiency | Autosomal recessive |
| SURF1 | Leigh syndrome SURF1-related | Autosomal recessive |
| SYN1 | X-linked epilepsy with variable learning disabilities | X-linked |
| SYNE4 | Autosomal recessive deafness 76 | Autosomal recessive |
| TAT | Tyrosinemia type II | Autosomal recessive |
| TAZ | Barth syndrome | X-linked |
| TBCE | Hypoparathyroidism-retardation-dysmorphism syndrome | Autosomal recessive |
| TBX19 | Adrenocorticotropic hormone deficiency | Autosomal recessive |
| TCIRG1 | Osteopetrosis 1 | Autosomal recessive |
| TCTN1 | Joubert syndrome 13 | Autosomal recessive |
| TCTN2 | TCTN2-related ciliopathies | Autosomal recessive |
| TCTN3 | Joubert syndrome 18 | Autosomal recessive |
| TECPR2 | Spastic paraplegia 49 | Autosomal recessive |
| TERT | Dyskeratosis congenita type 4 | Autosomal recessive |
| TF | Atransferrinemia | Autosomal recessive |
| TFR2 | Hemochromatosis type 3 | Autosomal recessive |
| TG | Thyroid dyshormonogenesis TG-related | Autosomal recessive |
| TGM1 | Congenital ichthyosis | Autosomal recessive |
| TH | Segawa syndrome | Autosomal recessive |
| THOC2 | X-linked Intellectual disability THOC2-related | X-linked |
| TK2 | Mitochondrial DNA depletion syndrome 2 | Autosomal recessive |
| TMC1 | Nonsyndromic hearing loss 7 | Autosomal recessive |
| TMEM138 | Joubert syndrome 16 | Autosomal recessive |
| TMEM216 | TMEM216-related ciliopathies | Autosomal recessive |
| TMEM231 | Joubert syndrome 20 | Autosomal recessive |
| TMEM237 | Joubert syndrome 14 | Autosomal recessive |
| TMEM38B | Osteogenesis imperfecta type XIV | Autosomal recessive |
| TMEM67 | COACH syndrome | Autosomal recessive |
| TMEM70 | Mitochondrial complex V deficiency type 2 | Autosomal recessive |
| TMPRSS3 | Nonsyndromic hearing loss TMPRSS3-related | Autosomal recessive |
| TNFSF11 | Osteopetrosis 2 | Autosomal recessive |
| TNXB | Ehlers–Danlos-like syndrome due to tenascin-X deficiency | Autosomal recessive |
| TPO | Thyroid dyshormonogenesis TPO-related | Autosomal recessive |
| TPP1 | Neuronal ceroid lipofuscinosis TPP1-related | Autosomal recessive |
| TRAPPC11 | Limb-girdle muscular dystrophy 18 | Autosomal recessive |
| TRDN | Catecholaminergic polymorphic ventricular tachycardia | Autosomal recessive |
| TREX1 | Aicardi-Goutieres syndrome 1 | Autosomal recessive |
| TRHR | Generalized thyrotropin-releasing hormone resistance | Autosomal recessive |
| TRIM32 | TRIM32-related disorders | Autosomal recessive |
| TRIM37 | Mulibrey nanism | Autosomal recessive |
| TRMU | Liver failure acute infantile | Autosomal recessive |
| TRPM6 | Hypomagnesemia 1 | Autosomal recessive |
| TSEN2 | Pontocerebellar hypoplasia type 2B | Autosomal recessive |
| TSEN34 | Pontocerebellar hypoplasia type 2C | Autosomal recessive |
| TSEN54 | Pontocerebellar hypoplasia type 2A | Autosomal recessive |
| TSFM | Combined oxidative phosphorylation deficiency TSFM-related | Autosomal recessive |
| TSHB | Congenital hypothyroidism TSHB-related | Autosomal recessive |
| TSHR | Congenital hypothyroidism TSHR-related | Autosomal recessive |
| TTC37 | Trichohepatoenteric syndrome | Autosomal recessive |
| TTC7A | Gastrointestinal defects and immunodeficiency syndrome | Autosomal recessive |
| TTC8 | Bardet-Biedl syndrome 8 | Autosomal recessive |
| TTPA | Ataxia with isolated vitamin E deficiency | Autosomal recessive |
| TULP1 | TULP1-related retinal disorders | Autosomal recessive |
| TYMP | Mitochondrial neurogastrointestinal encephalopathy (MNGIE) disease | Autosomal recessive |
| TYR | Oculocutaneous albinism types 1A and 1B | Autosomal recessive |
| TYRP1 | Oculocutaneous albinism type III | Autosomal recessive |
| UGT1A1 | Crigler-Najjar syndrome | Autosomal recessive |
| UNC13D | Familial hemophagocytic lymphohistiocytosis type 3 | Autosomal recessive |
| UPF3B | Lujan-Fryns syndrome UPF3B- related | X-linked |
| USH1C | USH1C-related disorders | Autosomal recessive |
| USH1G | Usher syndrome type IG | Autosomal recessive |
| USH2A | Usher syndrome type 2A | Autosomal recessive |
| VDR | Vitamin D-dependent rickets type 2A | Autosomal recessive |
| VLDLR | Cerebellar ataxia mental retardation and dysequilibrium syndrome 1 | Autosomal recessive |
| VPS13A | Choreoacanthocytosis | Autosomal recessive |
| VPS13B | Cohen syndrome | Autosomal recessive |
| VPS45 | Severe congenital neutropenia | Autosomal recessive |
| VPS53 | Pontocerebellar hypoplasia type 2E | Autosomal recessive |
| VRK1 | Pontocerebellar hypoplasia type 1A | Autosomal recessive |
| VSX2 | Microphthalmia with or without coloboma | Autosomal recessive |
| WAS | WAS-related hematopoietic disorder | X-linked |
| WHRN | Usher syndrome type 2D | Autosomal recessive |
| WISP3 | Progressive pseudorheumatoid dysplasia | Autosomal recessive |
| WNT1 | Osteogenesis imperfecta type 15 | Autosomal recessive |
| WNT10A | WNT10A-related ectodermal dysplasias | Autosomal recessive |
| WRN | Werner syndrome | Autosomal recessive |
| XPA | Xeroderma pigmentosum group A | Autosomal recessive |
| XPC | Xeroderma pigmentosum group C | Autosomal recessive |
| ZAP70 | ZAP70-related Immunodeficiency | Autosomal recessive |
| ZBTB24 | Immunodeficiency-centromeric instability-facial anomalies syndrome 2 | Autosomal recessive |
| ZDHHC9 | X-linked intellectual disability ZDHHC9-related | X-linked |
| ZFYVE26 | Spastic paraplegia 15 | Autosomal recessive |
| ZNF469 | Brittle cornea syndrome 1 | Autosomal recessive |
| ZNF711 | X-linked intellectual disability ZNF711-related | X-linked |